
Student Perspective: The role of the FDA in the future of medtech innovation
This op-ed is part of a series from E295: Communications for Engineering Leaders. In this course, Master of Engineering students were challenged to communicate a topic they found interesting to a broad audience of technical and non-technical readers.
Written by Kian Talaei, MEng '20 (IEOR)
Have you ever wondered how Theranos and its “revolutionary” technology slid past FDA regulations? New technology in general poses difficulties for regulators. With advances in artificial intelligence (AI) and machine learning (ML) in the medical technology industry (medtech), what will happen to FDA regulations in the near future?
Medtech is in a period of groundbreaking change, with data at the epicenter of that change. This change, however, brings a level of uncertainty for the FDA as they approach regulations. If the FDA is already encountering difficulties regulating “locked” AI/ML-based software as a medical device (SaMD), how will they handle adaptive SaMDs, whose algorithms “adapt” or change with new data introduced in the system? In the following, I introduce a regulatory solution to solve the huge challenge around regulating AI/ML-based innovations.
Theranos’s blood diagnosis machine was the first and biggest scandal in the dawn of advances in the medtech industry. How can a company raise so much money and survive so long without proper FDA backing? According to Elizabeth Holmes, FDA approval was only voluntary and her decision to receive FDA approval for the Herpes test was based on her decision that “FDA approval is an important step towards validating the company’s technology” (Friedman 2015). The FDA’s approach towards the Edison machine was highly unethical and nearly put the lives of millions in danger. The regulatory procedures towards the company in addition to the company’s misreporting of device capabilities should be a wake-up call for regulatory agencies to understand the necessary guidelines they need to uphold at all times.
Artificial intelligence, “the science of making intelligent machines,” and machine learning are revolutionizing many industries and are at the forefront of transformation in the medtech field (McCarthy 2017). Afshin Bazargan, a research and development director of Medtronic, stated how “artificial intelligence and machine learning and the data behind them are becoming extremely important in the medical device industry, especially in our approaches to concept generation. There is a new era of technology.” Within this new era of growth, there are two underlying regulatory distinctions to discuss. There are two types of AI/ML SaMDs: “locked” and “adaptive” (Babic et al 2019). Locked AI/ML-based SaMDs are those in which the algorithm for the device is invariable when sent for FDA approval; by contrast, adaptive refers to devices in which the algorithm “learns” based on new data. AI/ML-based SaMD is already assisting in early cancer detection using radiology images, early heart disease diagnosis using EKG data, and medication dosage selection using diagnostics and gene data (“Software as a Medical Device”). Based on the efficiency and impact of AI technology, our approach to healthcare will move towards a future of adaptive and data-driven medical devices (Figure 1).
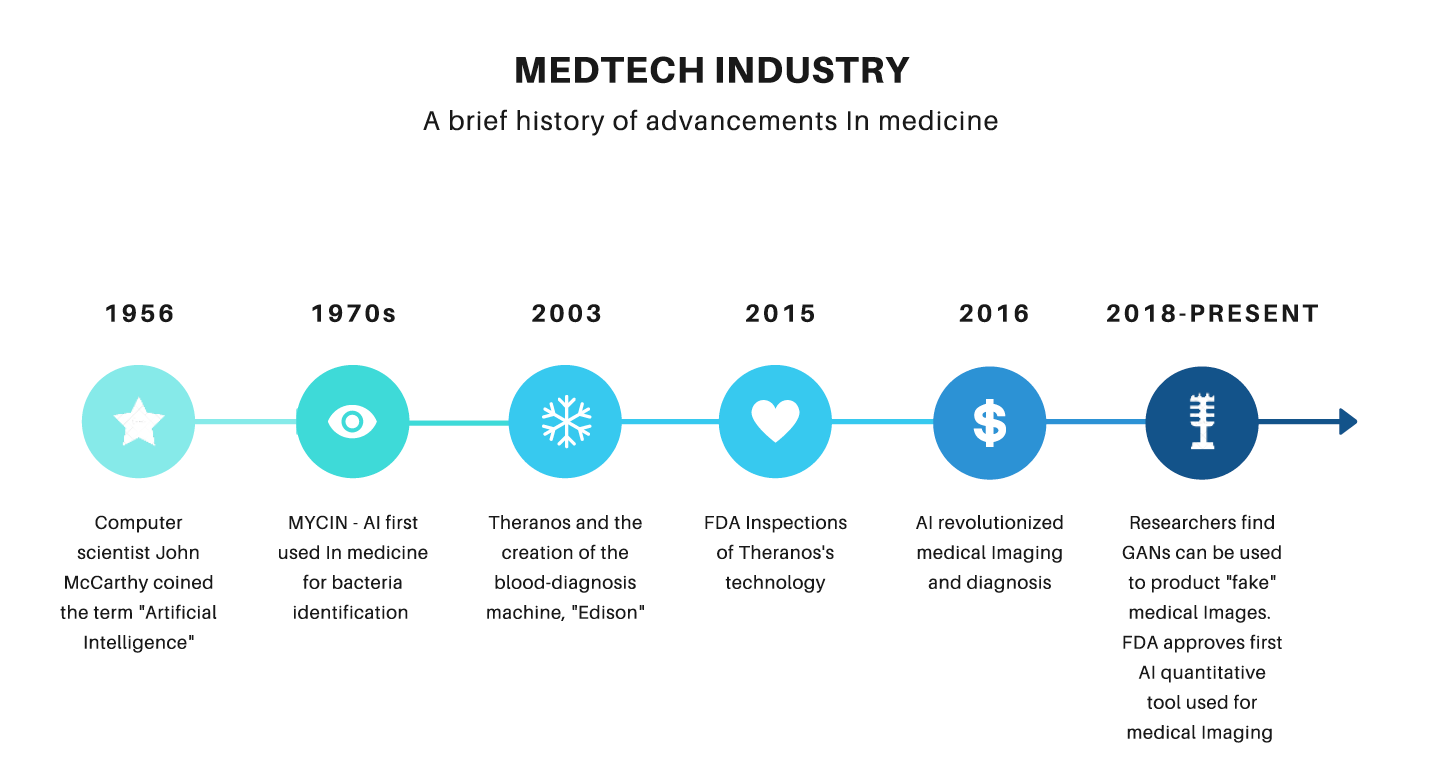
The FDA faces a new obstacle in tracking new advances in big data and artificial intelligence for devices. Nevertheless, that decision must keep one principle at its core: safety for the general public. As Mr. Bazargan puts it, “No matter how much the industry changes, the FDA can never and truly never loosen up on regulations. Enforcing strong clinical and research backing is crucial to the credibility of the industry.”
In other words, advances in technology must always prioritize public safety and privacy.
As depicted in Figure 2, current FDA regulations address locked SaMDs through regular pre-market approval, but adaptive devices, which frequently re-optimize to improve health, are not within the scope of current regulations (AI and ML in software 2019).

According to a recent article published by the FDA, a new adaptive device that can be used, for instance, to diagnose lung cancer growth must abide by four principles: good machine learning practices (GMLP), premarket assurance of safety and efficacy, review with established plan for modifications, and monitoring of product performance (Proposed Regulatory Framework 2019).
Let’s take one of these proposed regulations and dig deeper. Good machine learning practice refers to quality system excellence and “good software engineering practices” such as consistency in data acquisition and level of clarity. One of the biggest challenges this notion faces is advances in generative adversarial networks (GANs). Researchers at the University of Central Florida investigated whether radiologists can differentiate between “fake” visuals of lung cancer created using GANs and actual lung cancer diagnosis images (Chuquicusma et al. 2018). Their results showed that nearly all radiologists could not tell the difference between the images. This result opens doors into the problems the FDA can face if a company decides to send the FDA “fake” but consistent data with their innovation (Figure 1).
With safety at the core of the decision-making, what the FDA actually needs to focus on is the risk, monitoring, and privacy issues of these devices, not only pre-approval but post-approval as well. The pre-approval modifications are important in the regulations but play a smaller role in the overall impact they bring. Additionally, companies must play a part by running effective clinical trials, correctly modifying any technology, and being transparent with regulatory agencies. We need to focus our resources and energy on assuring that low-risk, continuously monitored, and adaptive devices can be utilized safely.
The world is changing as we know it. Technology will spread to every little corner of the world and soon we will be living in an era of autonomous cars, house-service bots, and automated medical devices. Out applications of AI and ML will transform the future of medicine. Sooner than later, medtech companies need to implement different adaptive algorithms that will change millions of lives. This rapid change, however, comes at a cost to regulatory agencies. How many FDA employees with strong medical expertise can vet complicated algorithms? With rapid change in the industry, who is assuring government regulators are up to speed with new technology? The FDA must effectively plan a guideline and workforce for managing adaptive medtech innovation and its built-in transformations.
***
Kian Talaei is a current Master of Engineering student at UC Berkeley, studying Industrial Engineering Operations Research (IEOR) with a focus on Data Analytics. Kian is an all-encompassing student interested in economics, medicine, and tech. Connect with Kian.
This article was originally posted at https://medium.com/the-coleman-fung-institute/op-ed-the-role-of-the-fda-in-the-future-of-medtech-innovation-fd3cbaadd6de
References:
- “Artificial Intelligence and Machine Learning in Software.” U.S. Food and Drug Administration, FDA, 2019.
- Chuquicusma, et al. “How to Fool Radiologists with Generative Adversarial Networks? A Visual Turing Test for Lung Cancer Diagnosis.” ArXiv.org, 9 Jan. 2018, arxiv.org/abs/1710.09762.
- “FDA Selects Participants for New Digital Health Software Precertification Pilot Program.” U.S. Food and Drug Administration, FDA, 2017.
- Friedman, Lauren F. “Controversial Multibillion-Dollar Health Startup Theranos Just Got a Huge Seal of Approval from the US Government.” Business Insider, Business Insider, 2 July 2015.
- McCarthy, John. “WHAT IS ARTIFICIAL INTELLIGENCE.” Stanford Computer Science , 12 Nov. 2017.
- “Proposed Regulatory Framework for Modifications to Artificial Intelligence/Machine Learning (AI/ML)-Based Software as a Medical Device.” U.S. Food and Drug Administration, 2019.
- “‘Software as a Medical Device’: Possible Framework for Risk Categorization and Corresponding Considerations.” IMDRF Software as a Medical Device (SaMD) Working Group, 18 Sept. 2014.
